cudaGSEA is a package for the efficient parallelization of Gene Set Enrichment Analysis (GSEA) using commonly available CUDA-enabled accelerators such as GPUs from the NVIDIA Geforce or NVIDIA Tesla series. See the corresponding paper at
http://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1244-x
for implementation details and benchmark results.
Just clone the repository, install the library and run the example code.
cd /tmp
git clone https://github.com/gravitino/cudaGSEA.git # clone this repository
cd cudaGSEA
sudo R CMD INSTALL cudaGSEA_1.0.0.tar.gz # install the library
Rscript example.R # run the example
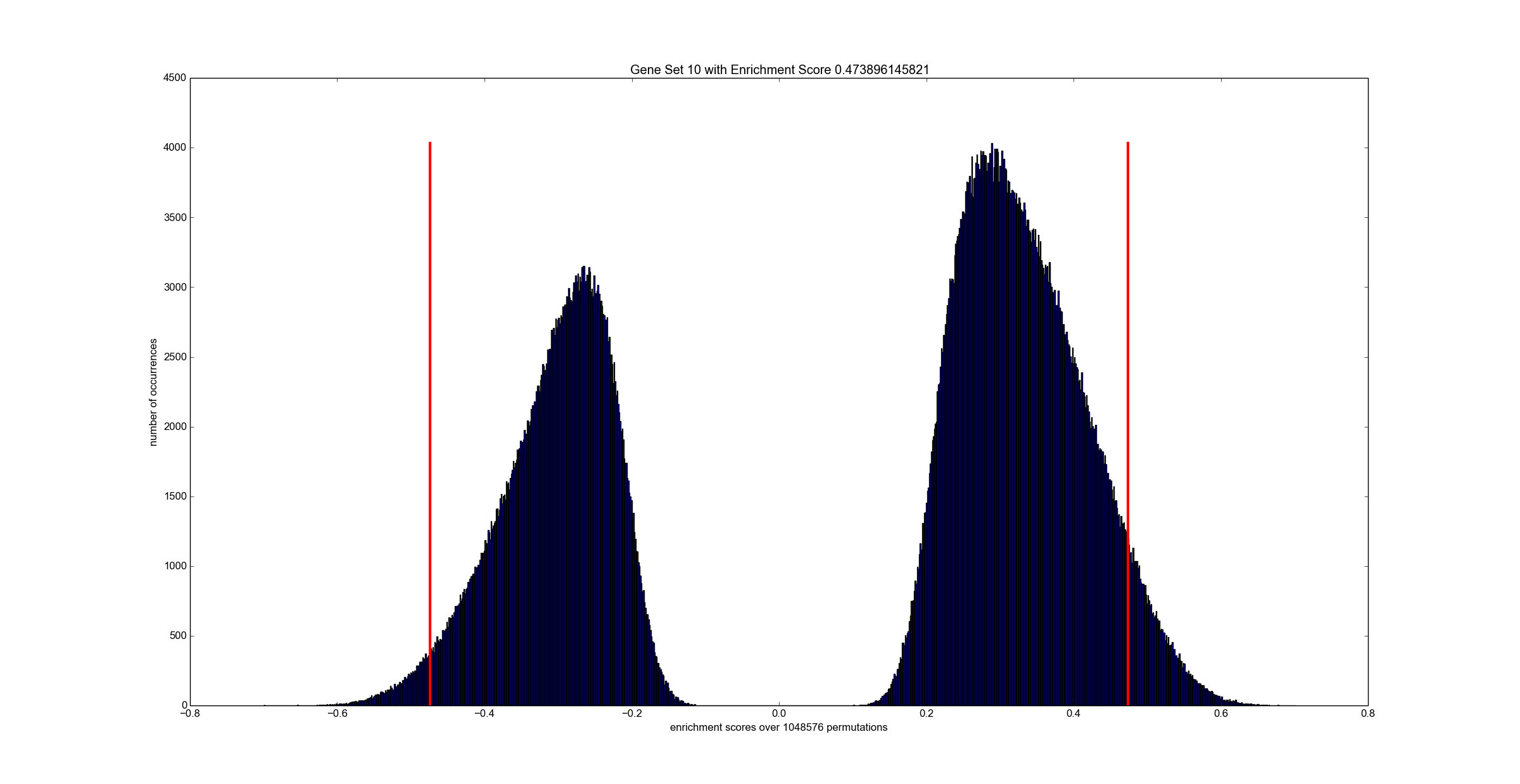
python tools/reader.py output_50_1024_32.es # optional: inspect all scores
The exemplary python visualizer needs numpy and matplotlib. However, you can use any other programming language since the output file is a simply binary dump of floating point values.
cudaGSEA depends on CUDA. Thus, you should at least be able to call the nvcc compiler from the command line before you try to install this package
-
Make sure you have a CUDA-enabled GPU with preferably lots of RAM, e.g. an NVIDIA Geforce GTX Titan X or an NVIDIA Tesla K40c. Older cards might also work but have not been tested.
-
Make sure you have installed CUDA 7.5 which can be obtained at https://developer.nvidia.com/cuda-downloads and gcc in version <5.0 (currently CUDA 7.5 does not support the newest gcc). As an example, Ubuntu 14.04 LTS comes with a correct version -- 15.10 does not! Currently, we are only supporting Linux.
-
You need R in version 3.2 and a current Rcpp package. Up to this point there are no dependencies on other Bioconductor packages such that cudaGSEA would also work as standalone library for plain R.
-
Get the package tarbal at https://github.com/gravitino/cudaGSEA
-
Install with "R CMD INSTALL cudaGSEA_1.0.0.tar.gz". This could take quite a bit since CUDA's nvcc is known to be not the fastest compiler.
The Makefile tries to automatically determine the correct paths for CUDA and R headers. This might not work properly if several CUDA or R versions are installed in parallel on your host system. In this case you have to explicitly set the correct paths to nvcc, R.h and Rcpp.h by altering the global environment variables:
-
export CUDA_HOME=/path/to/cuda-7.5 (directory of bin/nvcc)
-
export R_INC=/path/to/R/include (directory where R.hpp is located)
-
export RCPP_INC=/path/to/R/site-library/Rcpp/include (Rcpp.h)
If you encounter problems with libcudart please make sure your operating system knows how to find it. As an example, this is how you fix this issue on Ubuntu:
sudo echo "/usr/local/cuda-7.5/targets/x86_64-linux/lib/" >> \
/etc/ld.so.conf.d/cuda.conf # works on Ubuntu
sudo ldconfig
Do not hesitate to write me an email at [email protected]
cudaGSEA is designed to run on collapsed data i.e. genes have to be identified by appropriate gene symbols in order to work with gene sets from the MSigDB of the Broad Institute (http://software.broadinstitute.org/gsea/msigdb/index.jsp). Consequently, you have to manually collapse a raw gene expression data set with the chip file of the corresponding platform, e.g. by max pooling ambiguous array spots to a single and unique gene symbol.
cudaGSEA comes with three functions in order to configure the active GPU.
-
listCudaDevices() returns a list of CUDA-enabled accelerators.
-
setCudaDevice(deviceId) configures cudaGSEA to use the GPU with the provided device id. This is effectively a wrapper of the CUDA command cudaSetDevice(...).
-
getCudaDevice() returns the currently activated device. This is a wrapper to CUDA's cudaGetDevice() function. Note, if setCudaDevice uses an integer that is bigger than the amount of accessible GPUs then the device id defaults to 0.
If you want to test cudaGSEA on data stored in .gct, .cls and .gmt files you might use the three provided loading methods:
-
loadExpressionDataFromGCT("/path/to/expression/data/file.gct")
-
loadLabelsFromCLS("/path/to/label/data/file.cls")
-
loadGeneSetsFromGMT("/path/to/gene/sets/file.gmt")
The GSEA method takes five default arguments and three optional arguments
GSEA <- function(exprsData, labelList, geneSets, numPermutations, metricString,
dumpFileName="", checkInput=TRUE, doublePrecision=FALSE) {...}
exprsData, labelList and geneSets refer to the data obtained in the previous section. numPermutations denotes the number of permutations in the resampling test, metricString denotes the local ranking measure (one of the following):
- naive_diff_of_classes
- naive_ratio_of_classes
- naive_log2_ratio_of_classes
- stable_diff_of_classes
- stable_ratio_of_classes
- stable_log2_ratio_of_classes
- onepass_signal2noise
- onepass_t_test
- twopass_signal2noise
- twopass_t_test
- stable_signal2noise
- stable_t_test
- overkill_signal2noise
- overkill_t_test
dumpFileName specifies the name of a binary file which contains all enrichment scores for all permutations over all gene sets. The data is stored in a floating point array of "number of paths" x "number of permutations" many entries. See tools folder for a Python script that reads and visualizes enrichment scores. checkInput is a boolean flag that triggers sanity checks on the input -- keep this always activated unless you know what you are doing. doublePrecision triggers computation in double-precision. Note, single-precision is the default.
library(cudaGSEA) # the cudaGSEA library
# read data from cls, gmt and gct file formats
exprsData <- loadExpressionDataFromGCT("data/GSE19429/GSE19429_series.gct")
labelList <- loadLabelsFromCLS("data/GSE19429/GSE19429_series.cls")
geneSets <- loadGeneSetsFromGMT("data/Pathways/h.all.v5.0.symbols.gmt")
# access CUDA devices
listCudaDevices()
setCudaDevice(0)
getCudaDevice()
# configure GSEA
nperm <- 1024 # number of permutations
metric <- "onepass_signal2noise" # metric string see README.md
dump <- "" # if not empty path to binary dump
checkInput <-TRUE # check first three inputs or not
doublePrecision <-FALSE # compute in single or double prec.
GSEA(exprsData, labelList, geneSets, nperm, metric ,
dump, checkInput, doublePrecision)
Note, metrics on continuous data like Euclidean Distance and Pearson's Correlation Coefficient are not supported at the moment. The below stated metrics are two-class phenotype-based. Using default settings, standard deviations are biased (1/n) and adjusted for low values. Recompile with altered template settings to change behaviour. Further metrics may be added to (include/correlate_genes.cuh) using functors from (include/functors.cuh).
(computed with the following formula Mu(X) = E(X))
- naive_diff_of_classes
- naive_ratio_of_classes
- naive_log2_ratio_of_classes
(see Kahan, William (January 1965), "Further remarks on reducing truncation errors", Communications of the ACM 8 (1): 40, doi:10.1145/363707.363723)
- stable_diff_of_classes
- stable_ratio_of_classes
- stable_log2_ratio_of_classes
(computed with the following formula Var(X) = E(X^2)-E(X)^2)
- onepass_signal2noise
- onepass_t_test
(computed with the following formula Var(X) = E(X-E(X))^2)
- twopass_signal2noise
- twopass_t_test
(see Donald E. Knuth (1998). The Art of Computer Programming, volume 2: Seminumerical Algorithms, 3rd edn., p. 232. Boston: Addison-Wesley.)
- stable_signal2noise
- stable_t_test
(see http://i.stanford.edu/pub/cstr/reports/cs/tr/79/773/CS-TR-79-773.pdf)
- overkill_signal2noise
- overkill_t_test
All topics in this section cover source code related issues i.e. you have to alter the source code and reinstall the package!
Change your current directory to src and execute "make cudaGSEA".
See src/include/configuration.cuh and uncomment the corresponding defines.
See src/include/functors.cuh and alter or define new local measures
See src/include/error_codes.cuh .